10. Variational Circuit Preparation¶
10.1. Approximate ground states via tensor-network circuit optimization¶
We show how to represent an approximate ground state of an Hamiltonian expressed as Matrix-Product-Operator (MPO) as (i) a Matrix-Product-State (MPS), and as (ii) the wavefunction associated with a quantum circuit of finite depth. This reproduces (to some extent) the work of R. Haghshenas et al., Variational Power of Quantum Circuit Tensor Networks, Phys. Rev. X 12, 011047 (2022). This can be used to prepare an initial state for Quantum Phase Estimation (arxiv:2409.11748).
We also used some syntax and advice from the Tensor Network Training of Quantum Circuits example in \(\texttt{quimb}\)’s documentation.
import os
os.environ["OPENBLAS_NUM_THREADS"] = "1"
os.environ["JAX_ENABLE_X64"] = "True"
import autoray
import matplotlib.pyplot as plt
import numpy as np
import quimb.tensor as qtn
from qpe_toolbox.circuit import ansatz_circuit
from qpe_toolbox.estimation import qpe_sample, set_search_window
from qpe_toolbox.hamiltonian import heisenberg_hamiltonian
plt.rcParams.update({"font.size": 12})
opt = "auto-hq"
10.2. State preparation¶
10.2.1. The DMRG algorithm to find an MPS ground state¶
We first define our model Hamiltonian (Heisenberg chain) on \(n=8\) qubits. We know that its ground state can be approximated as a MPS. We use first the DMRG algorithm as a black box to have a reference energy (using a very large bond dimension 100 to have essentially the exact GS energy)
n_qubits = 8
hamilt = heisenberg_hamiltonian(n_qubits)
hamilt_mpo = hamilt.to_mpo()
hamilt_mpo.draw(
show_tags=True, show_inds=True, edge_scale=1, layout="kamada_kawai", edge_color=True
)
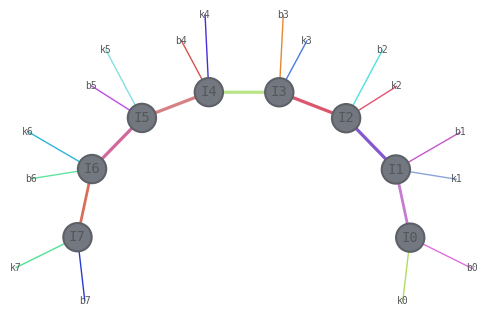
dmrg = qtn.DMRG2(hamilt_mpo, bond_dims=[10, 20, 40, 100], cutoffs=1e-13)
dmrg.solve(tol=1e-6, verbosity=1)
dmrg_energy = np.real(dmrg.energy)
print("DMRG reference energy:", dmrg_energy)
1, R, max_bond=(10/10), cutoff:1e-13
Energy: (-3.3749311611639135-3.3306690738754696e-16j) ... not converged.
2, R, max_bond=(10/20), cutoff:1e-13
Energy: (-3.374932598687866-2.220446049250313e-16j) ... not converged.
3, R, max_bond=(16/40), cutoff:1e-13
Energy: (-3.374932598687894-1.1102230246251565e-16j) ... converged!
DMRG reference energy: -3.374932598687894
10.2.2. MPS search via tensor-network differentiation¶
Let us now write a custom optimizer to find the ground state MPS, effectively performing the same task as in DMRG. The goal is simply to warm up for the circuit optimization that comes later.
When one optimizes the energy \(H\) with respect to a variational wave function, we need first to be able to evaluate \(\braket{\psi|H|\psi}\) as a tensor contraction
psi = qtn.MPS_rand_state(n_qubits, bond_dim=2)
psiH = psi.H
psi.align_(hamilt_mpo, psiH)
energy_tn = psiH & hamilt_mpo & psi
print("Contracted energy:", energy_tn.contract(all, optimize=opt))
energy_tn.draw()
Contracted energy: (0.5208302778006061+0j)
Now we can define a loss function, the energy, to be optimized w.r.t each tensor that makes the MPS \(\ket{\psi}\). We use \(\texttt{jax}\) as automatic differentiation tools to evaluate the gradients.
def loss(psi, mpo):
psiH = psi.H
norm_tn = psiH & psi
psi.align_(mpo, psiH)
energy_tn = psiH & mpo & psi
energy = autoray.do("real", energy_tn.contract(all, optimize=opt))
norm = autoray.do("real", norm_tn.contract(all, optimize=opt))
return energy / norm
def myoptimizer(chi, mpo):
psi = qtn.MPS_rand_state(n_qubits, bond_dim=chi)
return qtn.TNOptimizer(
psi, # the tensor network we want to optimize
loss, # the function we want to minimize
loss_constants={"mpo": mpo},
autodiff_backend="jax", # use 'autograd' for non-compiled optimization
optimizer="L-BFGS-B", # the optimization algorithm
)
Let us optimize for a first bond dimension \(\chi=4\). This is already extremely fast and accurate.
tn_opt = myoptimizer(4, hamilt_mpo)
psi_opt = tn_opt.optimize(n=200)
print("Optimized energy ", tn_opt.loss)
print("Exact energy ", dmrg_energy)
Optimized energy -3.3718017235650395
Exact energy -3.374932598687894
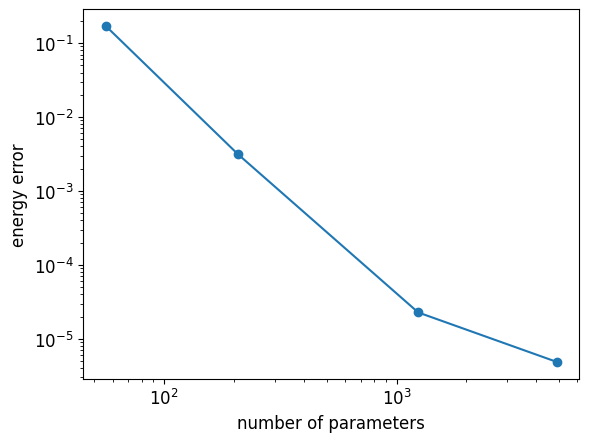
Let us be more quantitative by relating the energy estimation to the number of variational parameters \(d=2((n-2)\chi^2+2\chi)\) of an MPS, for various \(\chi\)
bond_dimensions = [2, 4, 10, 20]
n_values = len(bond_dimensions)
n_parameters = np.zeros(n_values)
optimized_energies = np.zeros(n_values)
for i in range(n_values):
tn_opt = myoptimizer(bond_dimensions[i], hamilt_mpo)
psi_opt = tn_opt.optimize(n=200)
n_parameters[i] = tn_opt.d
optimized_energies[i] = tn_opt.loss
plt.loglog(n_parameters, optimized_energies - dmrg_energy, "-o")
plt.xlabel("number of parameters")
plt.ylabel("energy error");
10.2.3. Finding a quantum circuit directly¶
MPS are good candidates for the initial state of the QPE algorithm because they can be prepared efficiently in a quantum computer. One possibility would be to find variationally the circuit that best approximates an MPS (see e.g. this recent proposal). Here, we take a shortcut, and directly find the circuit whose associated wavefunction minimizes the energy, in the spirit of R. Haghshenas et al., Variational Power of Quantum Circuit Tensor Networks. One practical advantage is that we may have less parameters to optimize using parametrized circuits compared to dense-tensors-based MPS.
We consider finite-depth quantum circuits made of parametrized \(U_3\) single qubit rotations and \(R_{ZZ}\) two qubit gates.
Let us visualize the circuit wavefunction, and the tensor network contraction associated with the estimation of the energy.
depth = 4
circ = ansatz_circuit(n_qubits, depth)
psi = circ.psi
psi.draw(color=["U3", "RZZ"], show_inds=True)
psiH = psi.H
psi.align_(hamilt_mpo, psiH)
energy_tn = psiH & hamilt_mpo & psi
print(len(energy_tn.tensors))
energy_tn.draw(color=["U3", "RZZ"], show_inds=True)
simplified_tn = energy_tn.full_simplify()
print(len(simplified_tn.tensors))
simplified_tn.draw(color=["U3", "RZZ"], show_inds=True)
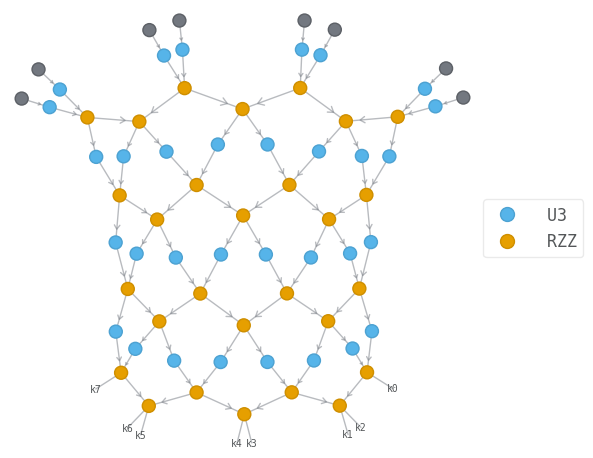
144
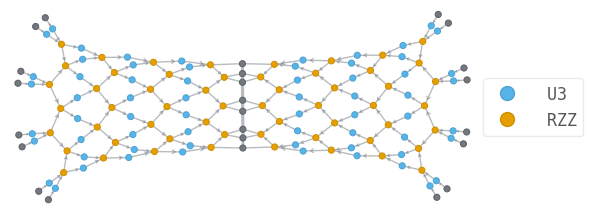
108
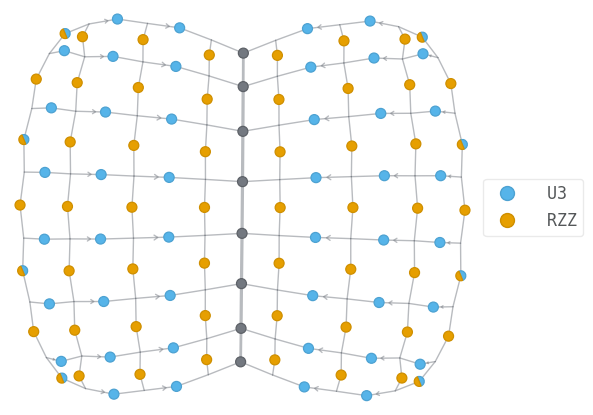
We can then define our optimizer on the angles that parametrize the gates.
def loss_circ(circ, mpo):
psi = circ.psi
psiH = psi.H
norm_tn = psiH & psi
psi.align_(mpo, psiH)
energy_tn = psiH & mpo & psi
energy = autoray.do("real", energy_tn.contract(all, optimize=opt))
norm = autoray.do("real", norm_tn.contract(all, optimize=opt))
return energy / norm
def make_circuit_optimizer(circ, mpo):
return qtn.TNOptimizer(
circ, # the tensor network we want to optimize
loss_circ, # the function we want to minimize
loss_constants={"mpo": mpo}, # supply U to the loss function as a constant TN
autodiff_backend="jax", # use 'autograd' for non-compiled optimization
optimizer="L-BFGS-B",
)
Let us make a first test. Since the contraction is a bit more involved than for straightforward MPS optimization, the simulation becomes a bit more tedious.
depth = 4
circ = ansatz_circuit(n_qubits, depth)
circ_optimizer = make_circuit_optimizer(circ, hamilt_mpo)
optimal_circuit = circ_optimizer.optimize(n=100)
print("Optimized energy ", circ_optimizer.loss)
print("Exact energy ", dmrg_energy)
circ_optimizer.plot();
Optimized energy -3.0927846148324294
Exact energy -3.374932598687894
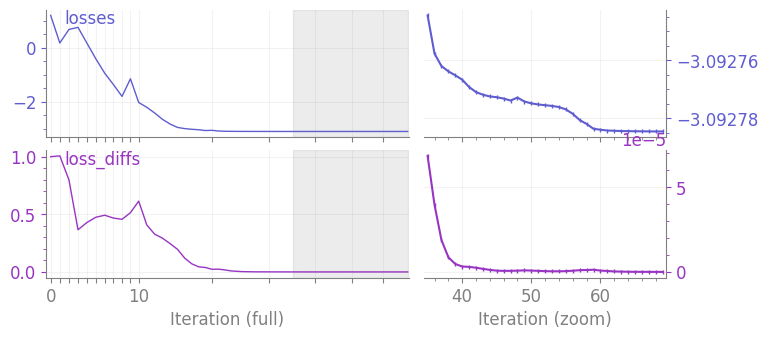
It can be also interesting to optimize a circuit of large depth using the optimized parameters from a circuit of smaller depth. However as shown below, we don’t observe an improvement. So we will not use that in the following.
# optimize a shallow circuit
circ = ansatz_circuit(n_qubits, 2)
circ_optimizer = make_circuit_optimizer(circ, hamilt_mpo)
optimal_circuit = circ_optimizer.optimize(n=100)
# initialize a deeper circuit with previously optimized parameters
circ = ansatz_circuit(n_qubits, 4, param_scaling=1e-4)
circ.set_params(optimal_circuit.get_params())
# optimize the deeper circuit
circ_optimizer = make_circuit_optimizer(circ, hamilt_mpo)
optimal_circuit = circ_optimizer.optimize(n=100)
print("Optimized energy ", circ_optimizer.loss)
print("Exact energy ", dmrg_energy)
Optimized energy -3.030776405464583
Exact energy -3.374932598687894
Let us analyze how things improve increasing the depth
max_depth = 4
depths = np.arange(1, max_depth + 1)
n_values = len(depths)
depth_parameters = np.zeros(n_values)
depth_energies = np.zeros(n_values)
for i in range(n_values):
circ = ansatz_circuit(n_qubits, depths[i])
circ_optimizer = make_circuit_optimizer(circ, hamilt_mpo)
optimal_circuit = circ_optimizer.optimize(n=100)
depth_parameters[i] = circ_optimizer.d
depth_energies[i] = circ_optimizer.loss
print("Depth ", depths[i])
print("Current energy ", depth_energies[i])
Depth 1
Current energy -1.249999999318702
Depth 2
Current energy -3.030776404868382
Depth 3
Current energy -3.190766520460821
Depth 4
Current energy -3.218978898175611
plt.loglog(depth_parameters, depth_energies - dmrg_energy, "-s", color="tab:orange")
plt.xlabel("number of parameters")
plt.ylabel("energy error")
plt.title(f"Optimizing {n_qubits} qubits");
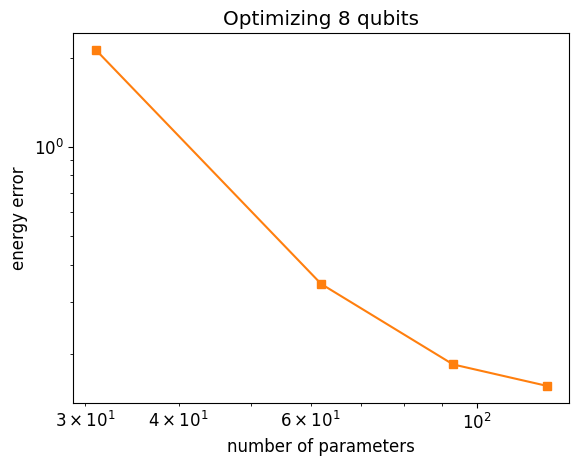
psi_exact = dmrg.state
overlap = (psi_exact.H & optimal_circuit.psi).contract()
print("fidelity ", abs(overlap) ** 2)
fidelity 0.8989035603516774
10.3. Quantum Phase Estimation¶
Let us know use a guess state the circuit optimization to initialize the QPE algorithm. For simplicity we will take a Hamiltonian defined on \(n=4\) qubits.
n_qubits = 4
hamilt = heisenberg_hamiltonian(n_qubits)
hamilt_mpo = hamilt.to_mpo()
dmrg = qtn.DMRG2(hamilt_mpo, bond_dims=[10, 20, 40, 100], cutoffs=1e-13)
dmrg.solve(tol=1e-6, verbosity=1)
dmrg_energy = np.real(dmrg.energy)
psi_exact = dmrg.state
print("\nStored reference energy ", dmrg_energy)
1, R, max_bond=(10/10), cutoff:1e-13
Energy: (-1.616025403784439-5.551115123125783e-17j) ... not converged.
2, R, max_bond=(4/20), cutoff:1e-13
Energy: (-1.6160254037844406+8.326672684688677e-17j) ... converged!
Stored reference energy -1.6160254037844406
We take the shallowest circuit
depth = 1
circ = ansatz_circuit(n_qubits, depth)
circ_optimizer = make_circuit_optimizer(circ, hamilt_mpo)
optimal_circuit = circ_optimizer.optimize(n=100)
print("Optimized energy ", circ_optimizer.loss)
print("Exact energy ", dmrg_energy)
Optimized energy -0.4999999996494705
Exact energy -1.6160254037844406
With this depth, we should be able to reach around 30% fidelity. If it isn’t the case, try to rerun the optimization
overlap = (psi_exact.H & optimal_circuit.psi).contract()
fidelity = abs(overlap) ** 2
print("fidelity ", fidelity)
fidelity 0.311004233762749
Running QPE on a state with 30% overlap should increase the fidelity. Let us initialize a circuit with a physical register and a phase register. We take \(m=3\) phase qubits.
n_phase_qubits = 3
# circuit from optimized gates
circ_qpe = qtn.CircuitMPS(n_phase_qubits + n_qubits)
for gate in optimal_circuit.gates:
label, params, qubits = gate.label, gate.params, gate.qubits
qubits = tuple(qb + n_phase_qubits for qb in qubits)
circ_qpe.apply_gate(label, *params, *qubits)
Set QPE parameters: first define the size of the search interval \(\Delta\) which sets the evolution time \(t\)
E_target = circ_optimizer.loss
size_interval = 3
_, evolution_time, global_phase = set_search_window(hamilt, E_target, size_interval)
and the Trotter parameters
trotter_order = 2
n_steps = 4
dt = evolution_time / n_steps
then run textbook QPE (this can take a minute or two). Since n_qubits is small, for faster execution you can always use exact time evolution instead of a Trotterization.
%%time
traces, res = qpe_sample(
hamilt, circ_qpe, evolution_time, dt, global_phase, trotter_order=trotter_order
)
CPU times: user 45.8 s, sys: 717 ms, total: 46.5 s
Wall time: 44.2 s
NB: the energy minimum can be \(< E_{\rm exact}\) when \(E_{\rm target} - \Delta/2 < E_{\rm exact}\). We only consider the bitstrings with probability \(> 4/\pi^2 F\) where \(F = |\langle{\psi}|\psi_{\rm exact}\rangle|^2\). The \(4/\pi^2\) factor gives a lower bound on the QPE success probability depending on the initial overlap as explained in the Textbook QPE example.
NB: in practice one will not have access to the overlap. An approximation of the fidelity is sufficient. The following quantity can be used as a proxy, see arxiv:2306.02620:
where \(\sigma = \langle H^2 \rangle - E^2\) is the energy variance on guess state \(\ket{\psi}\), and \(E_0\) is an estimate of the exact ground state energy that doesn’t need to be very accurate.
k_probs_list = sorted(enumerate(np.ravel(res)), key=lambda x: x[1], reverse=True)
kmin = 2 ** (n_phase_qubits - 1)
prob_min = 0
emin = E_target
energies = []
for k, prob in k_probs_list:
energy = E_target + size_interval * (1 / 2 - k / 2**n_phase_qubits)
energies.append(energy)
if energy < emin and prob > fidelity * 4 / np.pi**2:
emin = energy
kmin = k
prob_min = prob
print(f"Most probable energy = {energies[0]:.5f} w prob = {k_probs_list[0][1]:.5f}")
print(f"First 5 energies = {np.round(energies[:5], 5)}")
print(f'Minimal "significant" energy = {emin:.5f} with probability {prob_min:.5f}')
print(
f"error = {abs(dmrg_energy - emin):.5f} (theoretical error bound = {size_interval / 2**n_phase_qubits:.5f})"
)
Most probable energy = -0.12500 w prob = 0.33323
First 5 energies = [-0.125 -1.625 0.625 -0.5 0.25 ]
Minimal "significant" energy = -1.62500 with probability 0.32249
error = 0.00897 (theoretical error bound = 0.37500)
When the GS energy is measured, the physical register contains the GS wavefunction. We thus project the phase register of the final circuit wavefunction on the previously found bitstring that gives the minimal energy while satisfying the probability criterion
psi_final = traces["circuit"].psi
# change psi_final.backend from 'jax' to numpy as a workaround for
# https://github.com/jcmgray/quimb/issues/340
psi_final.apply_to_arrays(np.asarray)
bin_kmin = f"{kmin:b}".zfill(n_phase_qubits)
for i in range(n_phase_qubits):
psi_final.measure_(0, outcome=int(bin_kmin[i]), remove=True)
The fidelity should improve
overlap_qpe = psi_final.overlap(psi_exact)
fidelity_qpe = abs(overlap_qpe) ** 2
print(f"Fidelity before QPE = {fidelity:.5f}")
print(f"Fidelity after QPE = {fidelity_qpe:.5f}")
Fidelity before QPE = 0.31100
Fidelity after QPE = 0.96258
We observe that even with a moderate overlap, Quantum Phase Estimation projects on the ground state. As an exercise, you can try to reproduce the following figure:

it shows the infidelity as a function of circuit depth (orange curve) or bond dimension (blue curve) for a guess state prepared with either circuit optimization or DMRG. The colored stars then show how the fidelity quickly improves (error drops to \(\sim 10^{-5}\)) when running QPE with even a few phase qubits upon the trial state from circuit optimization.